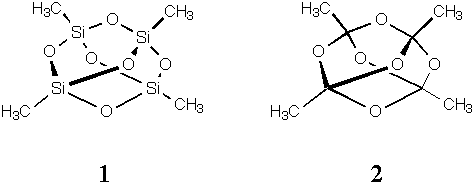
Using PC Model, answer the three questions below.
1. Energy minimize the structures of MeCuCN, MeAgCN, and MeAuCN. What are the three heavy atom bond lengths in each of these molecules? Do the structural results seem reasonable? Why or why not? In the breakdown of the MMX energies, you will note that many of the terms are exactly zero--why is that?
| Metal | r(M-CH3), Å | r(M-CN), Å | r(C-N), Å |
|---|---|---|---|
| Cu | 2.100 | 1.830 | 1.164 |
| Ag | 2.295 | 2.025 | 1.164 |
| Au | 2.295 | 2.025 | 1.164 |
2. When an amino acid is capped at its N-terminus with an acetyl group and its C terminus as an N-methyl amide, it is referred to as a dipeptide; i.e., the alanine dipeptide is drawn below as its fully extended conformer. In a non-polar environment (e.g., in the hydrophobic interior of a protein) the alanine dipeptide prefers to hydrogen bond one amide to the other to create a seven-membered ring. When this happens, the methyl group may either be disposed in a pseudoequatorial or pseudoaxial position (C7eq and C7ax conformations). These dipeptide ring conformers are equivalent to g turns found in protein secondary structure (g turns are possible connections between antiparallel b sheets). What are the relative energies of these three conformers assuming PC Model mimics a hydrophobic environment? Now consider the unnatural amino acid t-leucine (replace the methyl group in alanine with a t-butyl group). What are the relative energies of the three corresponding conformers of t-leucine? Based on your results, would you expect t-leucine to be more or less likely than alanine to be found at the corner of a g turn in an enzyme? (Note: be careful about hydrogen bonding in PC Model -- it's easy to accidentally turn it off!)
MMX (relative) energies, kcal/mol
| Residue | Extended | C7eq | C7ax |
|---|---|---|---|
| Ala | -8.2 (2.1) | -10.3 (0.0) | -9.2 (1.1) |
| t-Leu | -5.1 (0.9) | -6.0 (0.0) | -4.7 (1.3) |
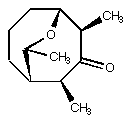
3. The natural product shown below inspires mayflies to stand
on their heads. It is thus an important target for total synthesis.
You have made it (optically pure, of course). On cooling your
NMR tube to -80° C, you observe two separate pairs of doublets
of quartets in the region of the 1H NMR corresponding
to protons alpha to a carbonyl. In one pair, the doublet splittings
are 1.1 and 3.7 Hz, respectively, while in the other pair, the
doublet splittings are 1.2 and 1.8 Hz, respectively (all quartet
splittings are about 7 Hz). Answer the following two questions:
(a) why are there two pairs of doublets of quartets? (b) what
is the integration ratio of one pair compared to the other (e.g.,
50:50, 80:20, etc?) Support your answer with data from PC Model.
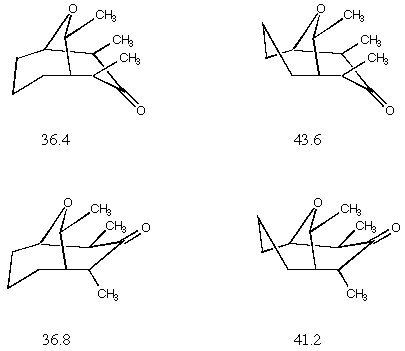
I found four stationary (i.e.,
all forces zero) conformers for this molecule which are illustrated
below in schematic fashion (the molecules are a bit too complex
for all-atom models to be sufficiently clear) The calculated MMX
energies are also listed.
As for the integration ratio, recalling
the formula for a Boltzmann distribution, the fraction of a conformer
A in an equilibrium mixture may be calculated as
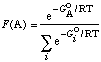 | (1) |
where the lower sum runs over all
equilibrium contributors (we'll make the approximation that the
free energy will be the same as the MMX energy). Using the energies
illustrated above and the experimental temperature of 193 K, one
calculates a ratio of the four isomers of 71:29:(5.7x10-07):(2.5x10-05).
Since your spectrometer isn't quite up to a 106
signal to noise ratio, you don't see the other two conformers.