Using PC Model, answer the three questions below.
1.The program will happily build polypeptides for you if properly instructed. What is the difference in energy between the a-helical form of Ala5 and it's fully extended (i.e., [[beta]]-sheet) form? What is the same difference for Gly5? Are your results consistent with the experimental observation that polyalanine forms a helices in water while polyglycine does not? One argument that might be leveled against comparing this calculation to experiment is that it is a gas-phase calculation. Recalculate the energy differences using a dielectric constant of 78.3 (water). How do things change? [Note: One could in principle worry a great deal about whether one should use the zwitterionic form of the polypeptide, whether one should try to optimize the torsions of the amino and carboxyl termini, etc. Don't. Just start from whatever PCModel draws (for goodness sake don't try to draw these by hand -- find the polypeptide construction menu!) and begin your minimizations from there.]
Heats of formation (kcal/mol) of various polypeptide conformers.
| dielectric=1.5 (default) | dielectric=78.3 | |||
| a-helix | b-sheet | a-helix | b-sheet | |
|---|---|---|---|---|
| Gly5 | -293.8 | -294.8 | -265.8 | -261.1 |
| Ala5 | -331.2 | -328.2 | -302.5 | -295.3 |
So, in the default dielectric of 1.5, polyalanine prefers the helix to the extended form, but polyglycine does not--this is entirely consistent with experimental observations. Raising the dielectric constant makes the heat of formation less negative in every case (because the dielectric is screening favorable interactions between charges) and now both polypeptides prefer helix over extended, although polyalanine does so to a larger extent (7.2 vs 4.7 kcal/mol). This illustrates the dangers of attempting to account for solvation with simply a bulk dielectric constant. The qualitative comparison between Ala and Gly is probably interpretable, but it is doubtful that the equilibrium constant between the two forms would be accurately calculated in any one of these instances. A very interesting question, however, is the accuracy of the qualitative observation that the preference for helix over extended conformation increases with increasing dielectric in these cases. This is probably correct, since a helix has a large net dipole, while the extended conformation does not.
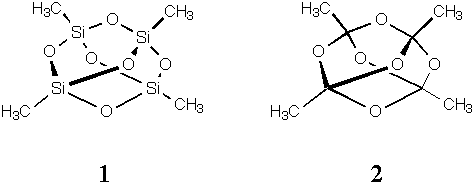
2.Minimize and report the heats of formation for structures 1 and 2. The former is methyl-terminated quartz, if you like, while the latter is a mildly unusual tetra-orthoester. One of these molecules has significantly more bending strain energy than the other. What linkage contributes most to this difference, that is, what angle has a lot more strain in one molecule compared to the analogous angle in the other molecule? You will need to support your answer either by comparing calculated angles to the tabulated equilibrium values in the force field (gotta find those, in that case) or by comparing to ostensibly strain-free molecules having the same linkage. To make life a bit simpler, let's assume all angles have the same force constant (i.e., only displacement from equilibrium matters here).
Hf(kcal/mol) -38.0 -226.7
The largest difference in angle strain (total 28.8 and 1.3 kcal/mol for 1 and 2, respectively) occurs for the XOX angle (where X = C or Si). In 1 this angle is 116.6o and in 2 this angle is 106.1o. In principle, the acyclic molecules H3SiOSiH3 and H3COCH3 are relatively unstrained and serve as useful models to examine the "preferred" XOX angle; it is 128.9o in H3SiOSiH3 and 111.8o in H3COCH3. Thus, this angle is distorted 12.3o in 1 and only 5.7o in 2. Since the angle bending energy is quadratic in displacement, this would be a roughly fourfold energy difference given identical force constants.
3.Cyclohexane is well known to have two minimum energy conformations, a chair and a twist-boat. PCModel predicts these to have heats of formation of -29.5 and -24.2 kcal/mol, respectively. This agrees closely with experiment (it was part of the parameterization of the force field, so that need not be surprising). Chair cyclohexane has D3h symmetry, the twist-boat has D2 symmetry--this implies that there is a single unique ring atom in the former, but in the latter there are two kinds of ring atoms. Tabulate the heats of formation for all possible stereoisomers that arise from substituting N, O, Si, P, or S for C (that is, do a complete conformational analysis for piperidine, tetrahydropyran, silacyclohexane, phosphorinane, and pentamethylene sulfide). Note that for the pnictogens, there may be issues associated with stereochemistry at that atom. In addition to the tabulation, comment on any particularly noteworthy trends in the energy differences (limit yourself to 100 words or less on your commentary).
By virtue of the symmetries of the species involved, there is only one chair form for all substitutions but N and P, where the pnictogen-H bond may be either axial or equatorial. The same is true of the twist boats with substitution of a heteroatom at one of the fourfold-equivalent positions. Substitution at the twofold-equivalent position delivers only one stereoisomer for any substitution since the hydrogen atoms on this position are chemically equivalent.
| X | |||||
| [H equatorial] | [H axial] | [H equatorial] | [H axial] | ||
| C | -29.5 | (-24.2) | |||
| N | -12.1 | -11.8 | -7.0 | -7.2 | -7.1 |
| O | -53.7 | -50.5 | -48.9 | ||
| Si | -24.3 | -20.2 | -18.9 | ||
| P | -20.8 | -22.6 | -17.3 | (not stationary) | -17.0 |
| S | -16.2 | -11.9 | -10.9 | ||
The relative energies of twist-boats vs chairs remains remarkably constant over all of these substitutions--about 5 kcal/mol. For most of the hetero-twist-boats, it is more favorable to put the heteroatom at the twofold-symmetric position than the fourfold-symmetric one. The conservation of relative energies is either wrong (bad force field?) or, if correct, is a coincidental cancellation of various effects (e.g., with O, there are no bad "flagpole" interactions across the twist boat with oxygen, but the C--O bonds are shorter than C--C, so the remaining interactions are worse than in cyclohexane).