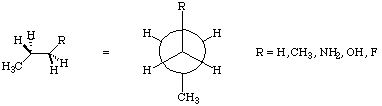
1) Recall that the molecular mechanics torsional potential may be expressed as:
Etorsion = 1/2 V1(1+cosw) + 1/2 V2(1+cos2w) + 1/2 V3(1+cos3w)
where the first term accounts for dipolar interactions, the second for hyperconjugation, and the third for bond-bond repulsions. Now consider the following series:
For each molecule in this series, use the Dihedral Driver under the Analyze menu to vary the dihedral angle (R-C-C-C) to construct a curve with (R-C-C-C) on the x-axis, and MMX energy on the y-axis. Collect enough data points to give a smooth curve which shows all regions of interest. Can you reconcile your graphs with the above equation, i.e. which term(s) seem(s) to dominate in each case? What is the barrier height for rotation in each case? Are there secondary barriers and minima? If so, what are their values? Does all of this seem chemically intuitive. Briefly state why or why not. Why is the MMX energy used for this exercise, rather than the heat of formation? Please attach printouts of your potential energy graphs.
This problem was designed to get you to think about the physical significance of each of the terms in the torsional potential. We are able to compare MMX energies for each series of rotamers, since we are comparing conformational isomers, i.e. all of the atom types are identical. In theory, we could also compare heats of formation. However, PCModel gives the exact same heat of formation for each rotamer, presumably because it doesn't recompute the heat of formation at each step. I'm not sure if this is a bug in PCModel (one of many, as you are now well aware), or a feature of the way the heat of formation is computed when a degree of freedom is fixed (the dihedral in this case). At any rate, the heats of formation are useless for the purpose of this excercise.
R=H: Your graph should be a perfectly symmetric cosine curve, with maxima at 0°, 120° and 240° and minima at 60°, 180° and 300°. This result indicates that the cos3w term dominates this rotational barrier. In fact, you can probably successfully model this rotation using only a cos3w term. I found the minimum to be 1.50 kcal/mol, and the maximum to be 4.54 kcal/mol, resulting in a barrier to rotation of 3.04 kcal/mol. This is consistent with experimentally derived barriers to C-C single bond rotation in similar molecules (2 - 3 kcal/mol), and very intuitive.
R=CH3: Your graph should now be symmetrically disposed with a global minimum at 180°, and lesser minima at 65° and 295°. The highest maximum is at 0°, with lower maxima at 120° and 240°. This appearance indicates a continued importance of the cos3w term, but it is no longer completely dominant. The dipolar cosw term is also important, and must be included to model this rotational barrier accurately. I found the values of the maxima to be 6.91 kcal/mol and 5.52 kcal/mol, while the minima are found at 3.07 kcal/mol and 2.17 kcal/mol. This gives barriers for partial rotation of ) 3.35 kcal/mol (anti to gauche), 3.84 kcal/mol (gauche to gauche) and 2.45 kcal/mol (gauche to anti). The anti to gauche rotation shows a higher barrier than to rotational barrier when R=H. This indicates that the dipolar term (cosw) has become important along with the bond-repulsion term (cos3w). The gauche to gauche rotation is the highest of the three. The increased barrier is likely due to two things; having to pass two bulky groups past each other (possibly accounted for in the cos3w term, but also accounted for in the van der Waals term, which we are not considering here) and increased hyperconjugative interactions (the cos2w term) in the gauche forms, which will concomitantly increase the stability of the gauche rotaomers while increasing the rotational barriers away from the gauche rotamers. Hyperconjugation is further evident in the displacement of the lesser minima from the expected 60° and 300°. The cos2w term, when turned on, will have exactly this effect (recall the minima for this term are at 90° and 270°). However, the hyperconjucation argument is seemingly not supported by the rotation barrier between gauche and anti, which is actually lower than that for R=H! This can be rationalized by considering the overall importance of the dipolar term (cosw). Incidentally, MMX predicts the gauche and anti forms of butane to differ by 0.9 kcal/mol, in exellent agreement with the experimental value of 0.97 kcal/mol. However, butane is used to provide the MM2/MMX parameters, so it better be right!!!
R=NH2: Qualitatively, this graph looks like that for R=CH3. My values for the maxima are 5.79 kcal/mol and 5.10 kcal/mol, with the minima having energies of 2.23 kcal/mol and 1.47 kcal/mol. Accordingly, the partial rotation barriers are 3.63 kcal/mol (anti to gauche), 3.56 kcal/mol (gauche to gauche), and 2.87 kcal/mol (gauche to anti). Again, all three terms play a roll. There is apparently an increased preference for dipolar alignment (C is more electronegative than N, so the N-C dipole should be stronger than the C-C dipole), as reflected by a slightly higher anti to gauche rotational barrier relative to R=CH3. However, the dipole from N-C should not lie directly on the axis of the bond, due to the lone pair, and should be opposite in direction to the C-C dipole, which makes this analysis less clear. The gauche to gauche barrier is reduced relative to R=CH3 (decreased steric repulsions?). Hyperconjugation may actually be more important in this case, as the gauche to anti rotational barrier is found to be higher. However, the lesser minima are not sigificantly displaced here, although there is observable asymmetry. One should keep in mind that we're trying to disect differences on the order of a few tenths of a kcal/mol. Such a difference is probably not meaningful at this level of theory.
R=OH: Again, this graph has a similar appearance to R=CH3 and R=NH2. I found values of 6.00 kcal/mol and 5.32 kcal/mol for the maxima, and 2.56 kcal/mol and 2.24 for the minima. The barriers are 3.08 kcal/mol (anti to gauche), 3.44 kcal/mol (gauche to gauche) and 2.76 kcal/mol (gauche to anti). These numbers are all smaller than those for R=NH2. Importantly, the gauche conformer is predicted to lie only 0.32 kcal/mol higher in energy than the anti conformer. Recalling that oxygen is more electronegative than carbon, one would expect the C-O bond dipole to flow towards oxygen, while the CH3-C dipole flows away from the methyl group. This means the anti conformer is actually worse in terms of dipolar alignment than the gauche conformers, so the cosw term has reduced importance in this system. Hyperconjugation also seems to be reduced, although this seems counterintuitive. The more electronegative oxygen should encourage electron flow from the hydrogens, increasing. Either this is not happening, or the effect is counterbalanced by some undetermined factor.
R=F: With R=F, the graph more closely resembles R=H than any of the others. This makes good sense, considering F is very close in size to H, and considerably smaller than all of the other ligands we considered. My energy values are 6.05 kcal/mol and 5.44 kcal/mol for the maxima, and 1.53 kcal/mol and 1.47 kcal/mol for the minima. Note the minima are now only 0.14 kcal/mol different in energy. The barriers for rotation are 4.00 kcal/mol (anti to gauche), 4.52 kcal/mol (gauche to gauche) and 3.91 kcal/mol (gauche to anti). Since F is extremely electronegative, hyperconjugation should be significant with both the methyl group and the hydrogens. The increased rotation barriers seem to support this. However, the graph shows less displacement in the lesser minima than for R=CH3. Therefore, if sterics are similar to R=H and hyperconjugation is not significantly reflected in the data, the dipolar term (cosw) must account for the high barrier to rotation. I am very dissatisfied with this result.
I was somewhat dissapointed with the results
from these calculations. I expected to see a clearer trend of
hyperconjucation becoming more important as the electronegativity
of the R group increased. Instead, the only data that reflect
significant weighting of the cos2w term is that for R=CH3.
For R=NH2, OH and F, the cos2w term is present, but
to a lesser degree than for R=CH3. For R=H, the cos2w
term seems to have a weighing of 0 (cosw term also?). Nevertheless,
with one or two expections, I feel that the results do make good
chemical sense. A more thourough study would have correlated
the CH2-CH2 bond lenth to rotational barrier,
and would have disected the total MMX energy into the torsional
term and the van der Waals term. However, that would have been
a lot more work, and I think I tortured you enough with this problem
set as it was!
2) Using the drawing tool, draw a hexagon. Use the bond tool (Add_B) to change every second bond from a single bond to a double bond. Click H/AD. You have just made cyclohexa-1,3,5-triene. Minimize this structure and make note of the MMX energy, heat of formation, and C-C bond lengths. Now, select every carbon and choose Piatoms from under the Mark menu. All of the carbons should now have a tilde (~) beside them. Re-minimize, and note the new data. Now, erase that structure, and choose Phen from the Rings menu. Minimize that structure, and note the same data. Which C6H6 structure has the lowest MMX energy (how much)? Which has the lowest heat of formation (how much)? Can you explain your observations? Why do the two types of energies seem to give different results? Repeat the first two steps for cycloocta-1,3,5,7-tetraene, and analyze the data. Can you draw any conclusions about p-delocalizaton, aromatic stabilization or anti-aromatic destabilization? Again, please be brief.
| C6H6 | C-C bond distance(s) | MMX energy | Heat of Formation |
|---|---|---|---|
| "triene" | 1.341 Å, 1.483 Å | 8.708 kcal/mol | 52.90 kcal/mol |
| "piatoms" | 1.372 Å, 1.433 Å | 8.914 kcal/mol | 22.87 kcal/mol |
| "aromatic" | 1.400 Å | 8.975 kcal/mol | 19.28 kcal/mol |
Interestingly, the MMX energy goes up as we improve our model of benzene, while the heat of formation goes down significantly. We can not compare MMX energies in this case because we are changing the atom types from one calculation to the next. We can compare heats of formation, since these species are structural isomers. I was able to coax the "piatom" result to match the "aromatic" result through repeated minimization, indicating that the "piatom" and "aromatic" atom types for carbon may actually be the same.
| C8H8 | C-C bond distances | MMX energy | Heat of Formation |
|---|---|---|---|
| PLANAR | |||
| "tetraene" | 1.337 Å, 1.479 Å | 38.068 | 96.19 |
| "piatoms" | 1.349 Å, 1.465 Å | 37.982 | 86.12 |
| BOAT | |||
| "tetraene" | 1.340 Å, 1.482 Å | 25.584 | 83.70 |
| "piatoms" | 1.345 Å, 1.486 Å | 27.775 | 70.65 |
Cyclooctatetraene (COT) is anti-aromatic, and actually exists in a boat conformation. If you chose the planar system, that's OK. The analysis is little effected by the choice of boat versus planar. However, note that the energies (either) are lower for the boat forms. I will continue my analysis using the boat form.
The original MM2 forcefield, on which MMX is built, parameterized Csp2-Csp2 interactions based on an isolated double bond, such as in 1-pentene. Consequently, MM2 gives lousy results for delocalized or aromatic systems. MMX attempts to fix this by including a correction that the user can turn on when delocalization is expected. For benzene, we see that the C-C bond lengths become much less alternant, and the heat of formation drops by 30 kcal/mol by turning delocalization on. Using the fully aromatic geometry, the heat of formation drops 3.5 kcal/mol.
Since COT is anti-aromatic, delocalization should be a non-factor. This is reflected in the very modest change in C-C bond lengths observed when delocalization is turned on. Nonetheless, the heat of formation drops by 7 kcal/mol when "piatoms" are used (10 kcal/mol for the planar system).
One may then conclude that any p delocalization is worth 7-10 kcal/mol, while aromatic stabilization gains an additional 20 kcal/mol. Experimental estimates for the aromatic stabilization in benzene range from 20-40 kcal/mol.
This question was not only designed to make you think about aromaticity, but was also conceived to drive home the point that atoms are not always what they seem. Picking the correct atom type is crucial to getting good molecular mechanics results.
3) Succinic acid [HOOC(CH2)2COOH] is a fairly floppy molecule. Minimize your initial structure and note the geometry and energies. By manipulating the atom positions and re-minimizing, what is the lowest energy form you can find? (I will base part of your points earned for this problem on how low your energy is relative to the structures found by the rest of the class.) Can you locate cyclic and/or hydrogen bonded structures? Report the lowest-energy "linear", cyclic (non-hydrogen bonded) and cyclic (hydrogen bonded) structures (one each is adequate; attach pictures if you wish). Can you rationalize the relative energies of the various minima you have located? Now, under the Options menu, set the dielectric constant to 78.3 (it was 1.5). This will very roughly approximate aqueous solution. Re-minimize your structures. Is the hydrogen bond retained? What is the lowest-energy solvated structure you can find? Compare the heats of formation for your entire ensemble (e = 1.5 and e = 78.3). Do you think it's valid to compare these numbers? Briefly justify your answers.
This excercise was largely designed to illustrate how many (local) energy minima exist for a molecule with many degrees of freedom. What you did, by picking a number of random starting geomtries for your minimizations, was a very limited Monte Carlo sampling of the potential energy hypersurface for succinic acid. If you had the type of luck I had, you probably found structures with heats of formation ranging from -180 kcal/mol to -192 kcal/mol (MMX energies perhaps as low as 0.1 kcal/mol). NOTE that if you neglected to hit H/AD before minimizing your structure, you may have tricked yourself into thinking that you found a negative MMX energy. In general, a molecular mechanics energy will not drop below zero, since zero indicates perfect agreement with the parameters. (It is mathematically possible, though...)
My lowest-energy structure (dielectric = 1.5) is an extended linear chain, with an MMX energy of 0.619 kcal/mol (heat of formation = -192.75). Any twisted, contorted or folded form I could find is at least 1 kcal/mol higher in energy. My hydrogen-bonded structure is 10 kcal/mol higher in energy. With "aqueous solution" turned on, the hydrogen bonded structure dissapeared. This is because the dielectric medium provides suitable interaction with the hydrogen bond donors and acceptors, while allowing the molecule to assume a more favorable conformation. In fact, I found that almost all of my "solvated" structures have heats of formation around -191 kcal/mol, indicating that the overall conformation of the molecule is less important than it's ability to expose its lone pairs and acidic protons to the dielectric medium.
Incidentally, I noticed that my hydroxide oxygens only had one lone pair when they should have two. Adding a second lone pair dramatically increases the energy. I suspect this is another bug in PCModel.
For this problem, it doesn't matter which energies you chose to compare. Everything should be comparable across the board, and in either dielectric, since we are again studying conformational isomers rather than strucutral isomers. Incidentally, there is no way to prove that the lowest energy conformer found by the class is the lowest energy conformer (i.e. the global minimum). Proving this would require the sampling of every single point on the hypersurface. Quite a daunting task...
4) Succinic acid forms a cyclic anhydride by simple heating:
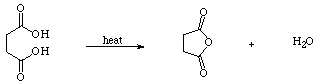
Use PCModel to estimate the barrier for this process, and plot a slice of the potential energy surface (PES) connecting reactant and products, and including any relevant intermediates and/or transition states. Do this with e = 1.5 and e = 78.3. Is the overall reaction endothermic or exothermic? What is the barrier? Do these numbers seem reasonable? (Hint: To plot a valid PES, your equation must always be balanced. For example, do not compare the heat of formation of succinic acid to the heat of formation of succinic anhydride. Instead, compare the heat of formation of succinic acid to the summed heats of formation of succinic anhydride and water.)
Phew. This problem was not meant to be such the exercise in futility that it turned out to be. I did want to illustrate the shortcomings of molecular mechanics for studying reaction pathways, but I didn't realize how significant the shortcoming of PCModel are.
There are a number of possible mechanisms for this process. I have picked the two most reasonable, and assumed that the pH is low enough (around 3) to keep everything protonated for simplicity's sake. The most theoretically sound mechanism involves backside attack of one of the carbonyl oxygens on the opposite carbonyl carbon. (This can be rationalized by MO theory and/or resonance structures. More on this in your next problem set...) The other mechanism I will consider is attack of the hydroxide oxygen in a similar manner. The validity of other mechanisms will be considered on a case-by-case basis.
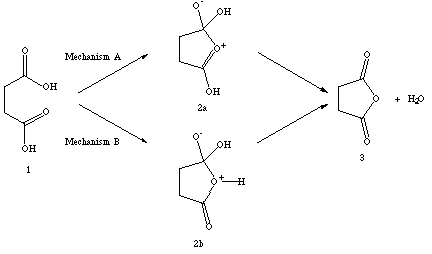
Regardless of which mechanism you chose, you started with succinic acid (1), and ended with succinic anhydride (3) and water. Since the atom types are changing during the course of the rearrangement, we must consider heats of formation. Recall that direct comparison of heats of formation may only be made for isomeric systems, so we must add the heats of formation of succinic anhydride and water for comparison to succinic acid. I used my lowest energy for 1 from problem #3, and found the sum of 3 + water to be -187.75 kcal/mol with e=1.5, and -186.99 kcal/mol with e=78.3. With these data points, I observed that the reaction is slightly endothermic (no more than 4 kcal/mol) at both dielectric settings. This is reasonable, considering the calculations are done at room temperature, and the reaction only procedes upon heating. Also consider that we are considering only the enthalpy, and not also the entropy (i.e., to be more correct, we should calculate the free energy change, not just the enthalpy change). Furthermore, it is more than possible that the calculated errors in the heats of formation are on this order of magnitude. So, I will conclude that this reaction is nearly thermoneutral, with a good chance of becoming spontaneous at increased temperatures.
Now, about the barrier... Plain and simple, there is no good way to depict the intermediate structure for either mechanism. MMX is not parameterized for enough oxonium atom types to allow minimzation of any proper structures. For my prefered mechanism (Mechanism A), one can draw a resonance structure that places the positive charge a carbon. While this maintains overall neutrality, the heat of formation is preposterously high (7.95 kcal/mol at e=1.5 and 60.73 kcal/mol at e=78.3). Using these numbers, the reaction barrier would be between 200 kcal/mol and 250 kcal/mol. Ludicrous!!
It is impossible to calculate an intermediate structure for the second mechanism which maintains charge neutrality. If one cheats, and uses a normal oxygen with one lone pair deleted rather than an oxonium ion, PCModel will compute a structure. Doing this, I found a barrier height of 20 kcal/mol in the "gas" phase, and 13 kcal/mol in the "aqueous" phase. These numbers are much more reasonable. However, I noticed that I had no lone pairs on the "oxonium" ion. There should be one. Playing around with this some, I discovered that I can get a wide variety of energies depending on how many lone pairs I placed on various oxygens. With the "correct" number of lone pairs on each oxygen, the calculated barrier returned to the 100+ kcal/mol realm. The net result is that PCModel does not know how many lone pairs should be on each oxygen, and that you can get just about any answer you desire. Very dissatisfying.
One final point. We have no way of knowing whether 2a (or 2b) is an intermediate or a transition state. If it is an intermediate, then there must be (higher energy) transition states between 1 and 2a, and 2a and 3 + 4. Therefore, our calculated barriers may only be lower limits to the actual barrier. To settle this issue, we would have to compute the infrared frequencies, which require second derivatives of the energy with respect to displacements. Such a calculation is impossible with PCModel, and most other molecular mechanics programs. We will explore this issue in more detail in forthcoming problem sets.