The Problems:
1. Use AM1 and PM3 to calculate the "heats of formation"
of the following molecules: F2, Cl2,
ClF, and ClF3. (i) Experimental heats of formation
are available for the first three molecules; locate the data and
decide which Hamiltonian is better for these calculations. Explain
your answer. (ii) An alternative method to evaluate the utility
of the methods is to calculate the enthalpy of reaction for F2
+ Cl2 --> 2 ClF. Based on this approach, which
Hamiltonian is better and why? (iii) So, armed with this analysis
and any other data you can find, comment on your ClF3
calculations.
2. DNA replication must proceed with extremely high fidelity in
order to minimize the possibly disastrous effects of random base-pair
mismatches (i.e., mutations). At left below is the standard Watson-Crick
CG base pair. One possible source of mutations during DNA replication
is matching to tautomeric forms of the bases that are present
at equilibrium. For instance, the imino tautomer of cytosine can
form a strongly hydrogen-bonded reverse-Watson-Crick pair with
"normal" C (at right below). The frequency of such point
mutations is presumably related to the relative population of
imino-C compared to C during replication. Using AM1, calculate
the relative energies of these two tautomers of cytosine (take
R as a methyl group in order to keep things simple-you do not
need to include the other base in this calculation). At 298 K,
what is the ratio of normal tautomer to imino tautomer (assuming
AM1 energies to be free energies)? To be viable, organisms need
roughly part per million to part per billion fidelity in their
DNA replication. Should you be panicking? Why or why not? Please
attach your .arc file for each tautomer
to your completed problem set.
Normal C_G base pair Abnormal Im C_C Base Pair
3. The Diels Alder reaction of chlorocyclopentadiene (ccp)
and acrylonitrile (an) is shown below. The cycloaddition
may proceed to give two regioisomeric products (for simplicity,
we're going to consider only these products of endo cycloaddition,
and ignore the two exo possibilities).
"ortho" "meta"
cycloadduct cycloadduct
pm3 tstate cycles=200 orthots.dat C 0.000000 0 0.000000 0 0.000000 0 0 0 0 -0.1579 C 1.407420 1 0.000000 0 0.000000 0 1 0 0 -0.1165 C 1.407388 1 108.222809 1 0.000000 0 2 1 0 -0.1337 C 1.401980 1 108.878497 1 0.655392 1 3 2 1 -0.0794 C 1.516806 1 107.421165 1 -19.325044 1 1 2 3 -0.0679 C 2.111649 1 98.126062 1 74.935865 1 1 2 3 -0.1074 C 2.192134 1 97.219388 1 -73.671052 1 4 3 2 -0.0540 H 1.090130 1 125.771391 1 176.630449 1 4 3 2 0.1145 C 1.424320 1 100.983452 1 -55.362859 1 7 4 3 -0.1074 H 1.107449 1 110.935221 1 -87.949048 1 5 1 2 0.0900 Cl 1.672221 1 124.240841 1 -171.008255 1 1 2 3 0.0721 H 1.089270 1 125.781035 1 165.511688 1 2 1 5 0.1304 H 1.103272 1 114.899884 1 151.455100 1 5 1 2 0.0835 H 1.091466 1 91.984616 1 168.985907 1 6 1 2 0.0878 H 1.089692 1 125.128040 1 -174.154754 1 3 2 1 0.1296 H 1.089675 1 97.550695 1 55.006343 1 6 1 2 0.0910 H 1.100839 1 91.146960 1 -170.046145 1 7 4 3 0.1149 Xx 5.000000 0 90.000000 0 180.000000 0 9 7 4 N 1.161237 1 89.492365 1 -179.267975 1 9 18 7 -0.0897 0 0.000000 0 0.000000 0 0.000000 0 0 0 0
Oversimplified, but nevertheless often useful, models have been
developed to predict regioselectivity in Diels Alder reactions
like this one. In particular, one begins by examining the highest
occupied and lowest unoccupied orbitals on the diene and dienophile
(i.e., HOMOs and LUMOs, which are nearly always the relevant pi
orbitals in these simple cases). One interaction occurs with a
smaller energy separation than the other, and is hence the most
relevant for a frontier orbital analysis. For a "standard"
Diels Alder reaction, it is the HOMO-diene/LUMO-dienophile interaction
(thus standard Diels Alder reactions are accelerated by pi donors
on the diene (raises HOMO energy) and pi acceptors on the dienophile
(lowers LUMO energy)). For an "inverse-electron-demand"
Diels Alder, it is the HOMO-dienophile/LUMO-diene separation that
is smallest. What are the HOMO and LUMO energies of ccp
and an? (You will need to include the VECTORS keyword in
your input file in order to have the molecular orbitals and their
energies printed.) Is this a standard Diels Alder reaction or
an inverse-electron-demand? Once you have the relevant HOMO and
LUMO, look at the orbital coefficients. Since the molecules are
planar, and the pi orbitals use only the out of plane p basis
functions, all of the coefficients in the HOMO and LUMO should
be zero (or very close to zero) except for the coefficients for
one kind of p function (probably pz). List
those coefficients for each atom in ccp and an.
The simplified models suggest that regioselectivity can be predicted
by comparing coefficients and matching the largest coefficient
for a diene terminus to the largest for the dienophile vinyl unit
(and, obviously, the smallest to the smallest). Does this work
in this case?
Appendix:
1.
| F2 | |||
| Cl2 | |||
| ClF | |||
| F2 + Cl2 -> 2 ClF | |||
| ClF3 |
This is not a problem designed to make you feel good about semiempirical
methods! Both AM1 and PM3 do awfully with the heats of formation
of gas-phase F2 and Cl2
(these are the standard states for these elements-hopefully you
didn't need to look up their heats of formation!) AM1 does OK
with ClF compared to PM3, but as a result the reaction enthalpy
for F2 + Cl2 ->
2 ClF is off by over 40 kcal/mol. PM3 is at least consistent
in its poor performance, and is off by a mere 16.5 kcal/mol. This
makes it a bit hard to decide which of AM1 or PM3 to trust more
for the heat of formation of ClF3. The fact
that they differ by 42+ kcal/mol is a bit disturbing! Read on
. . .
Structure of CF3:

In spite of the vastly different energies predicted for this molecule
by AM1 and PM3, they both predict similar structures, namely a
planar trigonal system with the bond lengths shown at left. What
a pity that experimentally the molecule is known to be T-shaped
as shown at right (can be found in any decent elementary inorganic
textbook). This problem represents a well known deficiency of
standard NDDO methods like AM1 and PM3-they do very poorly
with hypervalent molecules. Only the inclusion of d polarization
functions (available in semiempirical methods like MNDO/d and
PM3-TM) rectifies this problem.
2.
| Cytosine | |||
| Iminocytosine |
Wow! This implies that 6% of the time your DNA replicase is going
to do you in! Of course, you're not dead yet, which suggests that
either (i) AM1 gives a rotten equilibrium constant (That's probably
not the case. AM1 could certainly be off by as much as two orders
of magnitude in the equilibrium, but that would still be a far
cry from the part per million fidelity we're looking for.) (ii)
The gas-phase equilibrium is significantly affected by condensed
phase effects like solvation (a good possibility that is well
established for heterocycles-we'll come back to this issue in
the next problem set-keep your files.) (iii) Other enzymes check
for mismatches after the initial replication step and repair them
(this is typically true-take a biochem course if you want to know
more . . . )
3.
| Structure | |||
| acrylonitrile | |||
| chlorocyclopentadiene | |||
| Structure | |||
| ortho cycloadduct | |||
| meta cycloadduct | |||
| Structure | |||
| ortho TS structure | |||
| meta TS structure | |||
Obviously this is not the most regioselective Diels Alder reaction around . . . If you were hoping for meta adduct, you should run the reaction for a long time. If you want ortho adduct, you should stop it as soon as possible. Get ready for some unpleasant chromotography either way.
For the smallest HOMO-LUMO gap (label which orbital corresponds
to which molecule) list the pi coefficients (note that absolute
sign is arbitrary-only relative sign matters. That is, it doesn't
matter if your largest coefficient is negative on the acrylonitrile
vinyl unit but positive on a chlorocyclopentadiene terminus, those
are still the two centers that would be bonded to predict regioselectivity
using simple models).
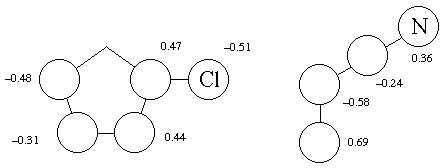
HOMO LUMO
Reaction is standard Diels Alder.
The coefficients don't offer any insight into the regioselectivity,
but then again, there isn't much kinetic regioselectivity. In
those cases where regioselectivity is high, this approach of analyzing
orbital coefficients works most of the time, but exceptions exist!
There is no substitute for simply calculating barrier heights,
which are expected to be reasonably good as far as relative
energies go, even at the semiempirical level.