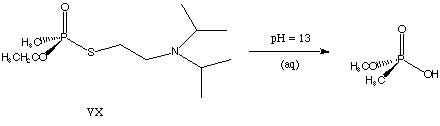
Prolog (or
"How Do I Run This Infernal Program?"):
1) Telnet over to the Cray C-90 (sk.networkcs.com). Hopefully, you picked this part up during the "Demo Amsol" day.
2) Your files must be named something.dat, where "something" is whatever you want to call that particular job, but the ".dat" is crucial. You will have to use the vi editor to either create a new input file from scratch, or to copy over an input file from SimpleText (or some other word-processor) on the Mac.
3) Once your files are in place, you're ready to roll! To run an AMSOL job, you will simply type amsolb something, where you must not include the ".dat" extension. If you do, the command will ask you to retype the input file name without the extension. Your job should now be submitted to the queue.
4) You may type qsr to see a list of all jobs running on the C-90. You should see yours. Once yours has disappeared from the list, it's done!
5) You may now look at your ".arc" and ".out" files with less something.out, or less something.arc. Type man less to get a help screen on less.
6) Several files may be deleted after your job has successfully completed. You may erase anything with ".com", ".qe" and ".qo" extensions (rm *.com, for example).
7) You only have about 1 hour of cpu time to last the entire quarter. Don't waste it! Type msiquery to see how many SUs you've used, and how many you have left.
8) If you have problems at any time, don't hesitate to ask for help. That's what Mike and I are here for.
9) The AMPAC and AMSOL manuals can be found in PDF format and in the Kolthoff computer lab.
1a) Construct z-matrices for the following molecules, taking full advantage of applicable symmetry:
molecular hydrogen (H2), acetylene, ethylene, ethane, benzene
Optimize each of these using the three NDDO Hamiltonians available in AMSOL (MNDO, AM1 and PM3).
Complete the following tables (or provide your own facsimiles). Experimental data which is not supplied may be found in any number of reference books. You will find the calculated heat of formation in the .arc file, on the line "Final heat of formation". If you have no .arc file for a given calculation, something went wrong, and you will have to look in the .out file for details.
Table 1.1: Heats of Formation, Computed and Experimental (kcal/mol)
| H2 | C2H2 | C2H4 | C2H6 | C6H6 | |
|---|---|---|---|---|---|
| MNDO | |||||
| Error | |||||
| AM1 | |||||
| Error | |||||
| PM3 | |||||
| Error | |||||
| Exp. |
Table 1.2: Enthalpies of Hydrogenation, Computed and Experimental (kcal/mol)
| H2 + C2H2 --> C2H4 | H2 + C2H4 --> C2H6 | |
|---|---|---|
| MNDO | ||
| Error | ||
| AM1 | ||
| Error | ||
| PM3 | ||
| Error | ||
| Exp. |
Which Hamiltonian provides the best agreement with experiment in Table 1.1? Is it the same in Table 1.2? Does this seem reasonable given what you know about the development history of semiempirical Hamiltonians?
b) Complete the following table. These distances can often be read from the z-matrix in the .arc file, which is the z-matrix for your optimized structure. The full interatomic distance matrix for your optimized structure can be found in the .out file, near the bottom.
Table 1.3: C-C Bond Lengths, Computed and Experimental (Å)
| C2H2 | C2H4 | C2H6 | C6H6 | |
|---|---|---|---|---|
| MNDO | ||||
| Error | ||||
| AM1 | ||||
| Error | ||||
| PM3 | ||||
| Error | ||||
| Exp. | 1.203 | 1.339 | 1.5351 | 1.399 |
a) Experimental data from the CRC handbook, 73rd Edition, "Bond Lengths and Angles in Gas-Phase Molecules", pp 9-15-9-41.
Which Hamiltonian gives the best agreement in each case in Table 1.3? Is there one Hamiltonian which seems to give the best overall agreement with experimental geometries? Which one?
Combining parts a) and b), can you identify one Hamiltonian that will always be best? If not, is there at least one or two that you think you could trust most of the time?
2) VX is an exceedingly nasty nerve agent. Most major militaries (including ours and Iraq's) have more than enough of this stuff on hand to wipe out every animal on this planet several times over. If someone drops some VX on your tank, and you're lucky enough to be wearing your protective gear, you're going to want to hop out and scrub your tank down before you rejoin the fight, just so you don't contaminate the rest of the battlefield. If you wash your tank with a basic aqueous solution, most of the VX will be neutralized, and you can get back to work. A reasonable guess at the first step for the neutralization reaction is:
MSDSs for VX and other nerve agents can be found at Military Unique MSDSs
If we want to study this process computationally, we can make our job easier by using a smaller model system for VX. This reduces conformational flexibility, size of our basis and computer time (recall cpu time is proportional to N4). Consider the following models for the above reaction.
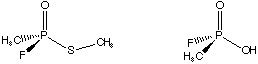
Build z-matrices for both of these (assume no symmetry, even though there is local symmetry in the methyl groups) and optimize them using the AM1 Hamiltonian. Don't worry about the stereochemistry. View your results with Chem3D and print pretty pictures for Mike to look at as he's grading. Do your structures look reasonable?
Now consider the following possible intermediate in this reaction. Note that the phosphorus in your input should be trigonal bipyramidal, with a 180° F-P-O(water) bond angle. Don't constrain this angle to be 180°, just be aware of this as you are writing your z-matrix. You might have to try a few times to find a z-matrix that works. Note that we don't have to concern ourselves with the charges on the atoms. Quantum theory knows how to treat that without any additional effort on our part.
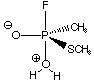
Attempt to optimize this structure, also at the AM1 level. Again, visualize the output, and attach a printout. Is the phosphorus still trigonal bipyramidal? Are there still five ligands within bonding distance of the phosphorus?
It is evident that phosphorus has a strong preference for one type of hybridization at the AM1 level. Explain this result.
3) Nikolai Menschutkin (1842-1907) was among the first to study solution effects on reactions which generate quaternary ammonium ions. As often happens in science, a prototypical reaction of this type bears his name:
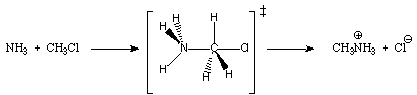
The Menschutkin reaction proceeds via a standard SN2 mechanism (see Abboud, J.-L. M.; Notario, R.; Bertrán, J.; Solà, M. "One Century of Physical Organic Chemistry: The Menschutkin Reaction" Prog. Phys. Org. Chem. 1993, 19, 1). Construct z-matrices for the reactants, transition state and products. Apply symmetry liberally. In the transition state, assume the hydrogens on the nitrogen are fixed staggered to the hydrogens on the carbon. Fix the N-C-Cl bond angle at 180° using a dummy atom. DO NOT fix the H(carbon)-C-Cl angle at 90°, i.e. DO NOT assume this structure is exactly trigonal bipyramidal at carbon. Using these guidelines, you should have little trouble finding this transition state.
We will run these calculations in the gas phase and in the aqueous phase using the SM5.4/P solvation model. The SM5 family does a very good job of accounting for bulk solvation effects. We will discuss solvation in more detail at the end of the quarter. If you want to know more now, refer to the AMSOL manual.
The following keywords should be used (again, refer to the manuals for details):
For neutral reactants:
PM3 SM5.4P SOLVNT=WATER TRUES SYMMETRY
For charged products:
PM3 SM5.4P SOLVNT=WATER TRUES SYMMETRY CHARGE=+1 (or -1 for chloride)
For the transition state:
PM3 SM5.4P SOLVNT=WATER TRUES SYMMETRY TSTATE
If your jobs completed successfully, you will have .arc files which now contain two sets of data. The top z-matrix and "Final heat of formation" are for the structure in the gas phase. The second z-matrix and "Heat of formation + Delta-G solvation" is for the structure optimized in water. Make a note of both energies.
a) Plot the gas phase potential energy surface (PES) for this reaction. What is the activation enthalpy? Is the overall reaction exothermic or endothermic? By how much?
Plot the solvated PES. How have the activation energy and thermicity of the reaction changed? Why has the PES changed so dramatically? (Hint: Recall that the gas-phase calculation is for an isolated molecule. The solvated calculation is still for one single molecule, but now it is surrounded by a polar medium. Evidence of the effect this has can be found by reading the "DeltaG-S(sol) free energy of solvation" line from the .arc file).
b) We have not done a complete job without confirming that the transition state structure really is a transition state for the reaction we want. Copy your transition state's .arc file to a new .dat file. Remove the first 29 lines, and edit the keywords to be:
PM3 FORCE THERMO ROT=3 TRANS=1
Look in the manual and tell me why ROT=3 and TRANS=1 have been chosen.
The interesting output from this job is in the .out file. Scan down the file until you find the "FREQUENCIES, REDUCED MASSES and VIBRATIONAL DIPOLES". What is your first frequency? Is it negative? Are there any other negative frequencies (ignore the last six printed)? A transition state must have one and only one negative frequency. If you find this in your output, congratulations, you found a transition state.
Scan down more to find "DESCRIPTION OF VIBRATIONS". What atomic motions are responsible for the negative frequency? Are these the motions you want to see for the transition state you were looking for? If not, try again. Otherwise, you are done. Please list the atoms and their shifts in your write-up, but you can ignore the percent radial movement.
Notes for your own enlightenment (these are not part of the assignment):
If we were going to publish this transition state, we would want to absolutely prove that it's the right one. Calculations can be performed which follow the downward curve of the PES. Called intrinsic reaction coordintate (IRC) following, or path following (slightly different from IRC), these calculations connect a transition state to the minima on either side in one smooth curve, allowing a plot of the true slice of the PES, rather than just three points. Incidentally, the output usually allows for easy animation of the PES. However, these calculations are difficult and take a lot of cpu time, so we won't do one here.
If we cared, we could use the thermodynamic data listed at the very end of the output file to compute thermal and entropic corrections to arrive at a true Gibbs' free energy. Again, we would do this if we wanted to publish our results, especially since the solvation energy is a free energy, so we ought to turn our heat of formation into a free energy to be proper about our comparisons. However, we don't really care at this point...
Note on Resources: All of these jobs are to be run in your account on the Cray C-90 (sk.networkcs.com). All you have to do is telnet over there to work. So, you can work from just about anywhere, including home if you have a modem and a SLIP/PPP account with the U. Chem3D is located in Kolthoff, so you will need to spend some time there to complete the assignment, unless you also have that at home.