| Chemistry 8003 | Computational Chemistry | 4 Credits | Winter Quarter 1999 | ( Due 3 / 8 / 99 ) |
A practical reminder: To run a job, you're input file should
be called myfile.dat. Simply issue
the command qrun myfile.dat and the
script will take care of the rest. If you want to look at an output
deck while a job is running, you can either vi
myfile.out or more myfile.out.
To examine the load leveler queue on the sp, the command is llq.
A nomenclature reminder: the notation x/y//w/z means level of
theory x using basis set y at a geometry optimized at level of
theory w with basis set z. E.g., MP4/6-311G**//HF/6-31G* means
the geometry was optimized at the HF/6-31G* level but the energy
(and/or other properties) are being calculated at the MP4/6-311G**
level.
Some quick notes/reminders with respect to Gaussian94:
1) Always include the keyword: scf=direct.
2) Always specify freq=noraman for
frequency calculations.
3) To find transition states in the absence of a symmetry
constraint, use fopt=(ts).
4) You can save a lot of time by using useful information
from previous calculations stored in the checkpoint file. Plan
your calculations to try to save time.
a) Keywords guess=read and geom=checkpoint
get the wave function and the geometry, respectively, from the
last completed calculation. So, if you have just done an optimization,
and want to follow-up with a frequency calculation, you will certainly
want to use these keywords. Note: Frequencies must be
calculated using the same level of theory with which the geometry
was optimized!
b) If you just optimized a geometry at one level of theory, and want to repeat that optimization at a different level, include readfc in the fopt=() keyword, e.g., fopt=(ts,readfc). This causes the program to start with the force constants from the previous calculation, which is efficient. It's usually a good idea to work your way up in a geometry optimization so you don't waste a lot of computer time because of a bad initial guess.
The Problems:
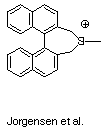
1. Last problem set, we examined the claims of Jorgensen et al. to have synthesized an isolated silicenium ion. More accurately, we showed why even arylsilicenium ions are highly unstable, raising doubts that an alkyl silicenium ion would be isolable. Let's now put the nails in the coffin. First, optimize the structure of tetramethylsilane (TMS) at the HF/STO-3G* level. Now, compute the NMR shielding of 29Si in TMS at the HF/321G*//HF/STO-3G* level (the keyword required is nmr). What is the absolute isotropic shielding in ppm? Now, compute the isotropic shielding of 29Si in PhSiH2+ at the HF/321G*//HF/STO-3G* level. Relative to TMS (which is the compound defining zero on the d scale for 29Si NMR) what is d for 29Si in PhSiH2+ (remember that d is relative de shielding)? Jorgensen et al. report a d value of 34 ppm for 29Si in their cation-does this seem possible given your calculations so far? Jorgensen et al. performed their experiments in acetonitrile (CH3CN). Compute the 29Si NMR d value for PhSiH2+ CH3CN. Is your value closer to that reported for the experimental system? Look at your structure for PhSiH2+ CH3CN; does it seem to be a true silicenium ion? Why or why not? If not, where is the positive charge localized? Should the Jorgensen et al. paper have been published?
2. We now return to the anomeric rotational potential studied
in both prior Problem Sets, where MMX and PM3 gave inconsistent
results. For the case of 2-methoxy-1,3-tetrahydropyran, find at
least one minimum energy structure and one transition state structure
at the RHF/3-21G level-provide pictures of your structures. Verify
the nature of these structures with frequency calculations. What
is the magnitude of the imaginary frequency for your TS structure?
Compute single-point energies at the B3LYP/631G*//HF/3-21G
level (use scf=(tight,direct) in single-point
keywords). What is your calculated rotational potential barrier?
Based on your ab initio calculations, was MMX or PM3 more accurate
for this molecule? Explain your answer.
3. Using your results from the above Problem 2, and assuming that
your minimum and barrier are unique on the torsional potential
energy surface, what does transition state theory predict the
rate constant for rotation to be at 298 K? (Recall TST says that
the rate constant for this unimolecular process is kBT/h
x exp(-DG‡/RT)
where kB is Boltzmann's constant, h is Planck's
constant, R is the universal gas constant, T is temperature, and
DG‡ is the
free energy of activation). To compute the free energies of your
species, use your B3LYP/631G*//HF/3-21G energies for potential
energy, and add the free energy contributions determined by your
frequency calculations at the HF/3-21G level. Now, compute the
aqueous solvation free energies for your two structures at the
SM5.42R/AM1 level using AMSOL (the keywords will be AM1 SM5.42R
1SCF SOLVNT=WATER TRUES HFCALC=1SCF). By what factor is solvation
predicted to affect the rotational rate constant (i.e., how does
the rate change when you modify DG‡(gas)
to instead be DG‡(aqueous)?)