| Chemistry 8003 | Computational Chemistry | 4 Credits | Winter Quarter 1999 | ( Due 3 / 8 / 99 ) |
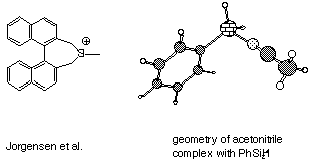
1. Last problem set, we examined the claims of Jorgensen et al.
to have synthesized an isolated silicenium ion. More accurately,
we showed why even arylsilicenium ions are highly unstable, raising
doubts that an alkyl silicenium ion would be isolable. Let's now
put the nails in the coffin. First, optimize the structure of
tetramethylsilane (TMS) at the HF/STO-3G* level. Now, compute
the NMR shielding of 29Si in TMS at the HF/321G*//HF/STO-3G*
level (the keyword required is nmr).
What is the absolute isotropic shielding in ppm? Now, compute
the isotropic shielding of 29Si in PhSiH2+
at the HF/321G*//HF/STO-3G* level. Relative to TMS (which
is the compound defining zero on the d
scale for 29Si NMR) what is d
for 29Si in PhSiH2+ (remember
that d is relative de shielding)?
Jorgensen et al. report a d value of
34 ppm for 29Si in their cation-does this
seem possible given your calculations so far? Jorgensen et al.
performed their experiments in acetonitrile (CH3CN).
Compute the 29Si NMR d
value for PhSiH2+ CH3CN.
Is your value closer to that reported for the experimental system?
Look at your structure for PhSiH2+ CH3CN;
does it seem to be a true silicenium ion? Why or why not? If not,
where is the positive charge localized? Should the Jorgensen et
al. paper have been published?
The absolute shielding of TMS (isotropic) is computed to be
514.2 ppm at the indicated level. At the same level, the d
values for PhSiH2+ and PhSiH2+
CH3CN are computed to be 218.4 and 12.1 ppm,
respectively. Only the latter is anywhere remotely close to the
experimentally reported value (the remaining discrepancy is not
unreasonable given the structural differences between our simple
aryl system and the real system-calculations on the real molecule
at a higher level of theory (too expensive for classwork) give
much better agreement). The acetonitrile complex has a normal,
covalent Si-N bond length. That is, there is zero silicenium
ion character. Instead, the cationic character is at the nitrile
carbon (the atomic partial charges bear this out as well). Indeed,
one might best view this as a methylated silyl isonitrile. Clearly,
the referees on the Jorgensen paper failed rather badly. Work
by Olah et al. is currently in press at JACS pointing out these
serious errors.
2. We now return to the anomeric rotational potential studied
in both prior Problem Sets, where MMX and PM3 gave inconsistent
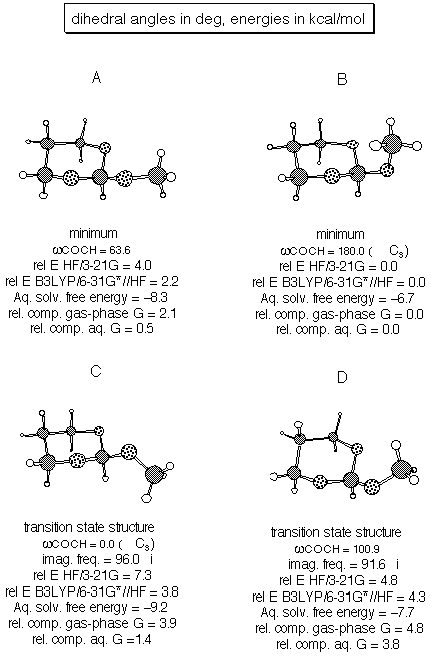
results. For the case of 2-methoxy-1,3-tetrahydropyran, find at
least one minimum energy structure and one transition state structure
at the RHF/3-21G level-provide pictures of your structures. Verify
the nature of these structures with frequency calculations. What
is the magnitude of the imaginary frequency for your TS structure?
Compute single-point energies at the B3LYP/631G*//HF/3-21G
level (use scf=(tight,direct) in single-point
keywords). What is your calculated rotational potential barrier?
Based on your ab initio calculations, was MMX or PM3 more accurate
for this molecule? Explain your answer.
Figures on the next two pages provide the critical data for this problem. Note how poorly RHF/3-21G does for relative energies compared to B3LYP/6-31G*. Note also how sensitive the aqueous solvation free energy is to conformation (the best solvated conformer is C, with all oxygen lone pairs pointing in the same direction for optimal solvation).
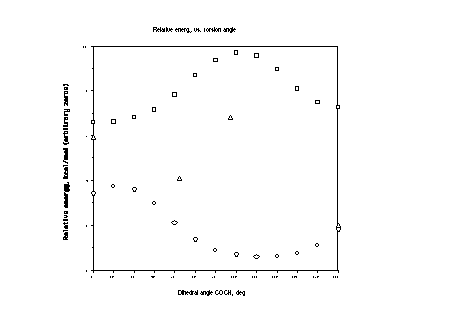
Above is the curve of relative energies, where squares are
PM3 and circles MMX and triangles B3LYP/6-31G*//HF/3-21G. I haven't
bothered to smooth the DFT curve. As to which level of theory
is better, MMX of PM3, the answer is somewhat depressing. Neither
does a great job, but PM3 is better. MMX has a minimum at zero,
where a local maximum on the coordinate should be, a maximum at
180, where a minimum should be, and completely misses the barrier
at 105 deg. PM3 properly has a minimum at 180, but fails to recognize
it as the global minimum. Also, PM3 has the secondary minimum
in the wrong place. In essence, PM3 fails to appreciate the full
magnitude of the anomeric effect, which should increasingly pull
the energy down as one travels from 0 to 180-or, in other words,
if we were to add a line of negative slope to the PM3 curve it
would reproduce the DFT curve reasonably well. So, the anomeric
effect is hard. Too bad, since we had such a nice rationalization
of our results for Problem Set 1 in terms of the anomeric effect.
This illustrates a general rule of theory: you can rationalize
anything, but you might want to avoid it until you're sure you're
prediction is correct!
3. Using your results from the above Problem 2, and assuming that
your minimum and barrier are unique on the torsional potential
energy surface, what does transition state theory predict the
rate constant for rotation to be at 298 K? (Recall TST says that
the rate constant for this unimolecular process is kBT/h
x exp(-DG‡/RT)
where kB is Boltzmann's constant, h is Planck's
constant, R is the universal gas constant, T is temperature, and
DG‡ is the
free energy of activation). To compute the free energies of your
species, use your B3LYP/631G*//HF/3-21G energies for potential
energy, and add the free energy contributions determined by your
frequency calculations at the HF/3-21G level. Now, compute the
aqueous solvation free energies for your two structures at the
SM5.42R/AM1 level using AMSOL (the keywords will be AM1 SM5.42R
1SCF SOLVNT=WATER TRUES HFCALC=1SCF). By what factor is solvation
predicted to affect the rotational rate constant (i.e., how does
the rate change when you modify DG‡(gas)
to instead be DG‡(aqueous)?)
The following data answer this question, which depends on which
minimum and which TS structure you found. Refer to pictures in
answer 2 for structures. Note how large an effect solvation can
have on the rate constant for even a fairly simple process like
bond rotation.