
Zhao, R.; Hettich, C.P.; Chen, X.; Gao, J.. npj Computational Material, 2021, 7, 148
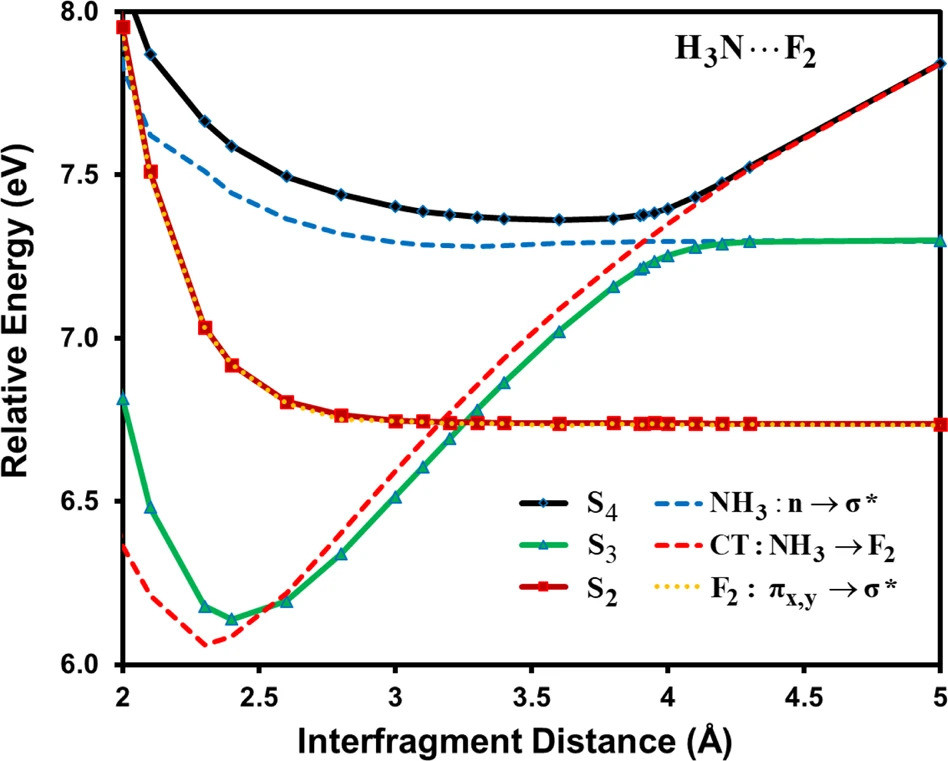
Multistate density functional theory (MSDFT) employing a minimum active space (MAS) is presented to determine charge transfer (CT) and local excited states of bimolecular complexes. MSDFT is a hybrid wave function theory (WFT) and density functional theory, in which dynamic correlation is first incorporated in individual determinant configurations using a Kohn–Sham exchange-correlation functional. Then, nonorthogonal configuration-state interaction is performed to treat static correlation. Because molecular orbitals are optimized separately for each determinant by including Kohn–Sham dynamic correlation, a minimal number of configurations in the active space, essential to representing low-lying excited and CT states of interest, is sufficient to yield the adiabatic states. We found that the present MAS-MSDFT method provides a good description of covalent and CT excited states in comparison with experiments and high-level computational results. Because of the simplicity and interpretive capability through diabatic configuration weights, the method may be useful in dynamic simulations of CT and nonadiabatic processes.