02/12/2008
Understanding Retention in Reversed-Phase Liquid Chromatography
Recent research from the group of Professor Ilja Siepmann.
Reversed-phase liquid chromatography (RPLC) is among the most widely
used and versatile analytical techniques. However, a detailed,
molecular-level understanding of the RPLC retention mechanism has
eluded analytical chemists for decades. Through validated,
particle-based Monte Carlo simulations of a model RPLC system
consisting of dimethyl octadecyl silanes on an explicit silica
substrate with unprotected residual silanols in contact with a
water/methanol mobile phase, graduate student
Jake Rafferty
and co-workers show that the molecular-level retention processes for
non-polar and polar analytes, such as alkanes and alcohols, are much
more complex than what has been previously deduced from thermodynamic
and theoretical arguments. In contrast to some previous assumptions,
the simulations indicate that both partitioning and adsorption play a
key role in the separation process and that the stationary phase in
RPLC behaves substantially different from a bulk hydrocarbon phase.
The retention of non-polar methylene segments is dominated by
lipophilic interactions with the retentive phase, while solvophilic
interactions are more important for the retention of the polar
hydroxyl group (see Figure).
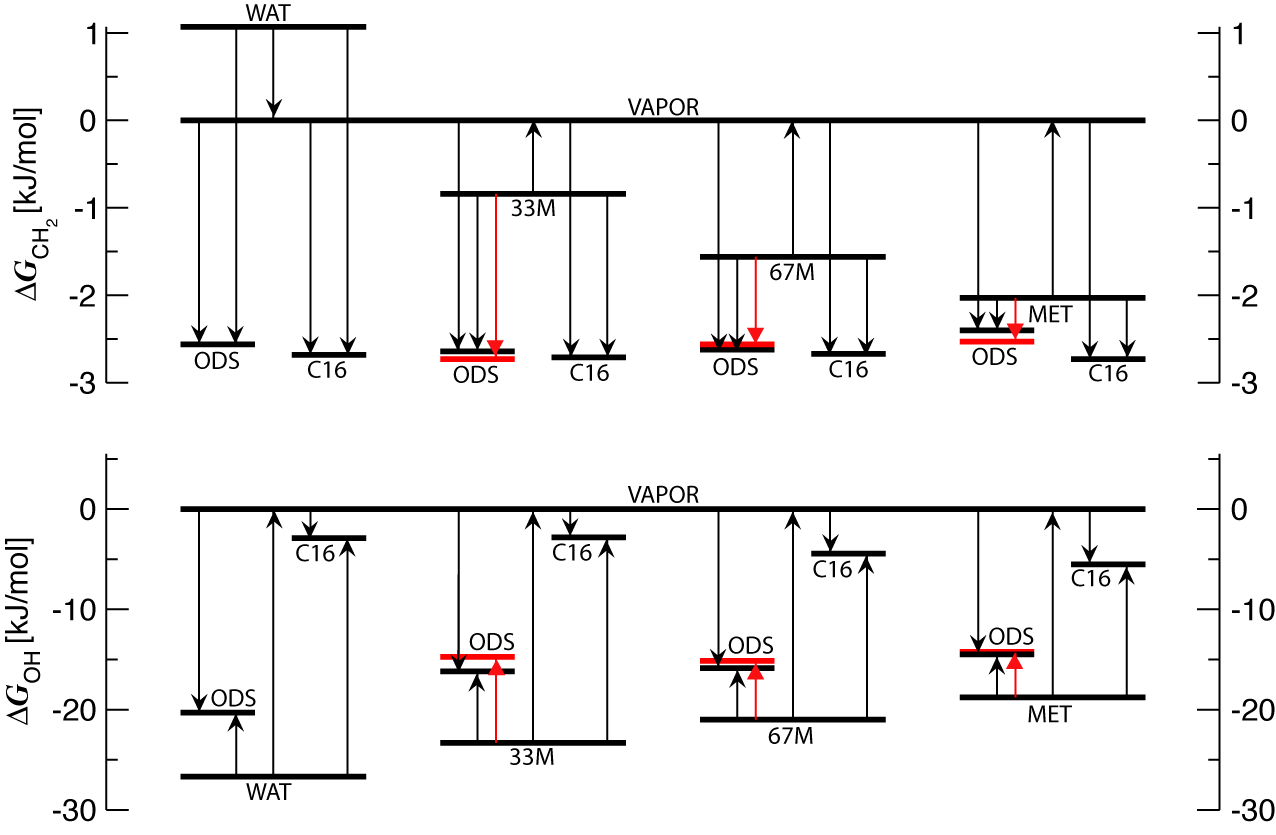
FIGURE CAPTION: Thermodynamic driving forces for retention. The
incremental free energies of transfer for methylene and hydroxyl
groups into the stationary phase (ODS) and mobile phases (neat water,
33% mole fraction of methanol, 67% mole fraction of methanol, and neat
methanol) are shown with respect to the ideal-gas reference phase
(VAPOR). For comparison, incremental free energies of transfer into
bulk n-hexadecane phase (C16) and determined from chromatographic
experiments (red lines, B.N. Barman, Ph.D. Dissertation, Georgetown
University, 1986) are also depicted.
A detailed description of this research has been published in
Analytical Chemistry 79, 6551 (2007).
The development of advanced computational strategies for the most
challenging problems in chemistry is a theme common to the research
endeavors of the
Minnesota Computational
Chemistry Group,
where research includes new theoretical formulations, the development
of new computational algorithms, and use of state-of-the-art
supercomputers to solve prototype problems to high accuracy and to
predict chemically useful results for a wide range of system scales
ranging from a few atoms to thousands of atoms.
Financial support from the National Science Foundation is gratefully
acknowledged. Part of the computer resources were provided by the
Minnesota Supercomputing Institute.
|