06/10/2008
Fe/Mn Swap in Dioxygenase Active Sites
Recent research from the group of Professor Lawrence Que.
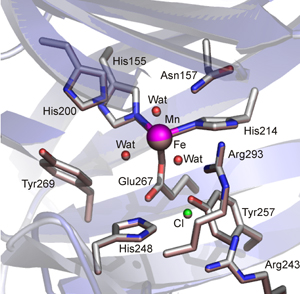
In work that appears in the May 27, 2008 issue
of Proceedings of the National Academy of Sciences USA (Proc. Natl.
Acad. Sci. USA 2008, 105, 7347-7352), postdoctoral
associates Joseph Emerson and Elena Kovaleva, together with graduate student
Erik Farquhar, in the groups of Profs. Larry Que and
John Lipscomb report
the surprising observation that the metal centers of a pair of iron- and
manganese-dependent dioxygenases, namely Fe-HPCD and Mn-MndD, can be swapped
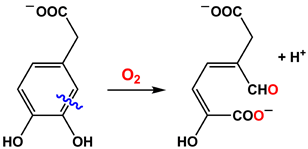
without affecting the catalytic parameters of these enzymes. Both enzymes
catalyze the activation of dioxygen to cleave the C2-C3 bond of 3,4-dihydroxyphenylacetate.
Fe-HPCD and Mn-MndD are respectively found in Brevibacterium
fuscum and Arthrobacter globiformis CM-2 and have been cloned
and over-expressed in E. coli. The two enzymes share >80% sequence
identity and have crystal structures that show nearly superimposable active
sites. Growth of the E. coli cells with the desired metal ion present
in excess allowed the isolation of Fe-MndD and Mn-HPCD, enzymes with the
non-native metal ion in the respective active sites. Interestingly, all
four enzyme preparations exhibited very
similar KM and Vmax values,
representing the first pair of structurally characterized metalloenzymes
that activate O2 and remain fully active even after a metal
swap. The near-superimposability of the two active sites suggests that
the protein environment does not tune the dramatically different redox
properties of Fe and Mn to achieve a common redox potential to carry out
oxygen activation. This remarkable observation leads us to conclude that
an integral redox change at the metal, specifically M(II) to M(III), is
not an essential feature of the catalytic mechanism, making this pair of
enzymes an exception to the widely accepted paradigm for oxygen activation
at transition metal centers. |