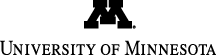
Main navigation | Main content

High-valent oxoiron species are implicated as oxidants for many iron enzymes that activate dioxygen. In the past ten years, Regents Professor Lawrence Que Jr.'s laboratory has focused on synthesizing oxoiron(IV) complexes that can serve as models for such enzyme intermediates, and a number of such complexes has been characterized by a variety of spectroscopic methods. Some have even been crystallized and their crystal structures have been solved. The latest developments in this effort have been reported in two papers recently published in Proceedings of the National Academy of Sciences (PNAS).
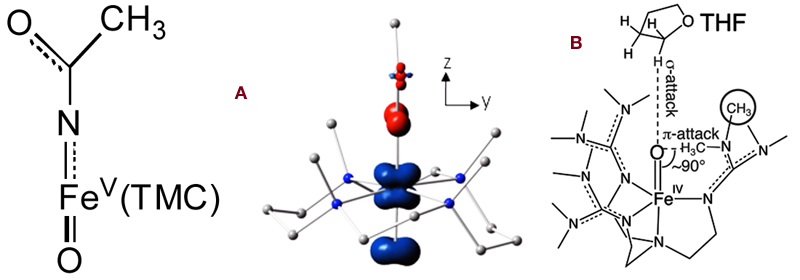
In the July 24 issue (PNAS 2012, 109, 11933), the synthesis of the first iron(V) complex with both oxo and imido ligands was reported, and its structure was deduced from a combination of UV-vis, resonance Raman, Mössbauer, EPR and mass spectral data. It is only the second oxoiron(V) complex to be characterized and is unique in having a neutral tetramethylcyclam (TMC) supporting ligand. By contrast, the first example has a tetraanionic macrocylic ligand to support and stabilize the oxoiron(V) unit. Thus, in the present case, the stabilization of the iron(V) state derives from the trans oxo and imido ligands. This paper is the culmination of efforts of three postdoctoral associates, Xiaopeng Shan, Adam Fiedler, and Kathy Van Heuvelen, and collaborators at Carnegie Mellon, Professors Eckard Münck and Emile Bominaar, Raymond De Hont, and Katlyn Meier. Graphic A
In the September 4 issue (PNAS 2012, 109, 14326), magnetic circular dichroism (MCD) studies on [FeIV(O)(TMG3tren)]2+ were reported in collaboration with the group of Professor Edward Solomon at Stanford University. This first example of a high-spin oxoiron(IV) complex to be characterized by X-ray crystallography serves to model corresponding transient intermediates trapped in enzymes by rapid-freeze-quench methods. [FeIV(O)(TMG3tren)]2+ can carry out intermolecular attack of the C-H bonds of added substrates, as well as intramolecular attack of a methyl group on the supporting tripodal ligand. This article presents detailed spectroscopic and high-level computational studies on the S = 2 FeIV=O center that define the nature of its frontier molecular orbitals (FMOs) and rationalize its reactivity in cleaving C-H bonds. Interestingly, because of limited access to the Fe=O unit, attack of an external substrate must occur along the Fe=O axis and involve the σ* FMO. On the other hand, the constraints imposed by the ligand architecture allow intramolecular attack of a ligand methyl group to occur only perpendicular to the Fe=O unit and must involve a π* FMO. Importantly, it is found that both π * and σ * reaction trajectories have comparable activation barriers and, in both cases, the FeIV=O unit acquires significant oxyl character, i.e. more FeIII-O•-like, as it elongates along the reaction coordinate towards the transition state. This difference in orientation may account for the different reactivities associated with the S = 2 FeIV=O unit in different enzyme active sites. Graphic B