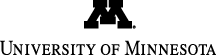
Main navigation | Main content

It is rare to be able to trap and identify a reactive intermediate associated with iron-catalyzed oxidations, but postdoctoral associate Williamson Oloo, Ph.D., in Professor Lawrence Que Jr.'s group has accomplished just such a feat, which was reported in Nature Communications (DOI: 10.1038/ncomms4046). This bio-inspired iron oxidation system consisting of a combination of a nonheme iron catalyst, H2O2, and acetic acid can carry out oxidations of C–H bonds in complex organic molecules with predictable regio- and stereoselectivity, as demonstrated by Christina White for (+)-artemisinin and (-)-dihydropleuromutilone, as well as highly enantioselective olefin epoxidation reactions as shown recently by Miquel Costas.
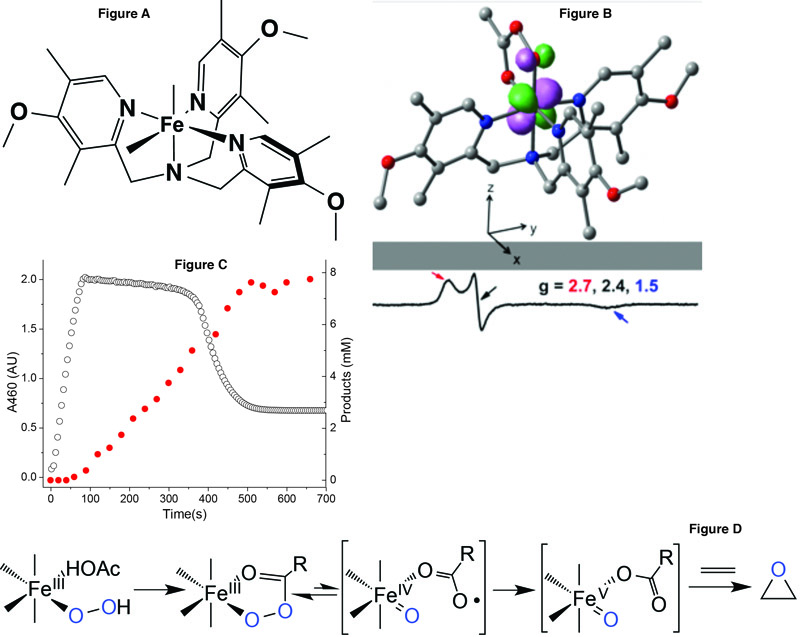
[See Figure A & B] Que's research group and others have invested considerable effort toward understanding the factors that contribute to the high efficiency and selectivity of this system. The researchers' interest was piqued by a report from Evgenii Talsi, who discovered an unusual EPR signal at g = 2.7 in the catalytic mixture that he attributed to an oxoiron(V) species. As this signal represented only about 10% of the iron in the sample, a higher concentration of the g = 2.7 species was desirable in order to be able to ascertain this assignment by the application of other spectroscopic methods. By using a more electron donating analog of the supporting tetradentate ligand and tweaking the reaction conditions, Oloo found that the fraction of the g = 2.7 intermediate in the samples could be increased to ~50%, which allowed its reliable characterization by a combination of UV-Vis, EPR, resonance Raman, Mössbauer spectroscopy (in collaboration with Katlyn Meier and Professor Eckard Münck at Carnegie Mellon University), and electrospray ionization mass spectrometry [See Figure C]. Together, these methods show that the g = 2.7 intermediate is NOT an oxoiron(V) species, as previously proposed, but is in fact a low-spin acylperoxoiron(III) species. By following its visible chromophore at 460 nm (open circles) as a function of time after the iron catalyst is mixed with H2O2 and acetic acid (AcOH) at -40 °C, this intermediate is found to form rapidly and to persist in a steady state phase during which catalytic olefin epoxidation occurs (filled circles). It then decays exponentially upon depletion of the H2O2, and epoxidation then stops. These results show that the g = 2.7 intermediate is not the actual epoxidizing agent but its precursor, which decays via a unimolecular rate-determining step to unmask a powerful oxidant. Density functional theory calculations showed that the acylperoxoiron(III) intermediate undergoes reversible O–O bond cleavage to form a FeIV(O)(•OAc) species. The latter then isomerizes to FeV(O)(OAc) that can oxidize substrate without a barrier. Researchers postulate that the enhanced catalytic activity and selectivity observed by the introduction of AcOH results from the involvement of the Fe(III)-OOAc intermediate and its ultimate conversion to a highly reactive FeV(O)(OAc) oxidant. [See Figure D]